학교, 연구소, 스타트업 창업, 회사 등 나름 다양한 환경을 겪어오면서 느낀 점. '생명정보학 인력'에 대한 정의와 기대 수준은 정말 천차만별 이라는 점. 오늘 이 혼란을 좀 정리해 보고자 한다.
크게 '생명정보학 인력'은 생명정보학 과학자와 생명정보학 엔지니어로 구분해 볼 수 있을 것 같다. 간략하게 정리해 보면, 아래와 같다. ( 각각의 기준에서의 최소 요건 )
Showing posts with label Bioinformatics. Show all posts
Showing posts with label Bioinformatics. Show all posts
Friday, June 17, 2016
Saturday, November 17, 2012
인간게놈프로젝트(Human Genome project)와 생물정보학(Bioinformatics)
Bioinformatics가 언론의 통해 기사화 되고 널리 알려지기 시작한 계기는 Genome Project 의 막바지인 1990년대 후반이 되면서 부터 였다. 인간게놈프로젝트( HGP: Human Genome Project)라 알려진 이 프로젝트는 10년의 시간에 걸쳐 미국 주도의 다국적 연구 컨소시움에 의해 총 30억달러 우리돈 3조원 이상이 소모된 달탐사 프로젝트 이래 최대의 과학 프로젝트 였다.
하지만, 1990년대 후반 크레이그 벤터( J. Craig Venter )가 셀레라(Celera genomics)사를 설립 대규모 자동 게놈 해독기를 장착하고 shortgun sequencing이라는 초고속 게놈 해독법을 이용해 인간 게놈을 분석하면서 이 지루한 프로젝트는 흥미진진한 소설 같은 상황으로 전개가 된다.
셀레라의 분석 기술로는 정부 주도의 다국적 팀에 의해 10년 이상이 소요될 것으로 예측되는 프로젝트를 불과 2-3년 안에 완료 할 수 있었고, 미생물 게놈을 빠른 시간 안에 분석 해 발표하면서 이는 기정 사실화 되었다. 정부 주도 하의 다국적 팀에게 이는 엄청난 위협이었고, 위기감을 불러 일으켰다. ( 과학자의 입장에서 보자면 수년간 노력한 일이 수포로 돌아가는 것과 마찬가지인데, 과학 연구에서 2등은 큰 의미가 없기 때문. 이와 관련한 스토리는 벤터의 자서전 Life decoded에 자세히 기록되어 있는데, 정부 주도 팀의 관료적 행태들은 구역질이 날 정도의 장면들이 많다. )
Shortgun genome sequencing
Shortgun sequencing 은 인간의 긴 30억개 염기 서열을 평균 500-1,000 개 염기 길이의 작은 단위로 잘라 읽은 후, 각각을 퍼즐 맞추기 식으로 이어 붙여 전체 30억개 염기 서열 부위를 완성하는 형태다.
Shotgun sequencing 방법은 얼핏 매우 간단해 보이지만, 이 방법은 Bioinformatics 가 없이는 애초에 불가능한 서열 분석 방법이었다.
Shortgun sequencing을 통해 예를 들어 세개의 염기 서열 조각이 아래와 같이 주어진다고 하면 어떻게 이어 붙여야 할까?
AGTGT, GTAAC, ACTAG
아래와 같이 이어 붙이면 된다.
AGTGT
GTAAC
ACTAG
--------------------------
AGTGTAACTAG
아래와 같은 방식도 가능하다.
ACTAG
AGTGT
GTAAC
--------------------------
ACTAGTGTAAC
어떤 쪽이 옳은 것일까? 이런 서열 조각이 수천만개 있다면 어떨까? 어떻게 이 조각들을 제대로 꿰어 맞출 수 있을까? 이는 굉장히 정교한 수학적 모델을 필요로 했으며, 엄청난 컴퓨팅 파워를 필요로 하는 문제로 정통 생물학자들이 해결할 수 있는 문제가 아니었다.
이 문제를 푸는데 적용할 만한 수학적 이론은 Shortest common superstring problem (SSP), Eulerian graph problem 등이 있었고, Genome sequencing에 적용할 만한 이론들로 Michael Waterman 등의 기여로 대체로 정립,개발되어 왔지만, 실제로 이를 대량의 염기 서열 조각을 이어 붙이는 시스템에 적용하여 제작된 전례가 없었다.
생물정보학자 전쟁 :: Eugene Myers vs. Jim Kent
Eugene Myers 를 필두로 하는 Bioinformatics 팀이 셀레라 측의 Shortgun sequencing을 위한 염기 서열 조각 맞추기 문제를 위한 시스템을 성공적으로 완성했다. Gene Myers라고 흔히 불리는 Eugene Myers 는 NCBI에서 1990년 지금까지 가장 광범위하고 성공적으로 사용되는 생물학 프로그램 BLAST 를 공동 개발한 Bioinformatics 초기 영웅 중 한명이었는데, 그가 Celera에 합류하여 라떼 커피를 친구삼아 밤낮 가리지 않고 일에 몰두한 덕에 비소로 셀레라의 Shortgun sequencing 기술이 완성될 수 있었다.
Shortgun sequencing 시스템을 하드웨어적, 소프트웨어적으로 완벽히 갖추고 인간 게놈 분석의 막바지에 다다르고 있었던 셀레라 사에 비해, 다국적팀( 인간게놈컨소시움)은 정부의 중재로 셀레라 사와 함께 인간 게놈 분석 초안을 발표하기로 약속한 몇달 전이 되기 까지 이 게놈 조각 퍼즐 맞추기( Sequence assembly ) 를 위한 시스템이 완성되지 않아 초조하게 발을 구르는 상황이었다.
10년의 노력, 3조원의 정부 연구비를 정당화 하기 위해서 이 시스템은 너무나 간절했던 것이었다. 이 때, UCSC 생물학과에서 박사과정 학생이던 Jim Kent (William James Kent )가 David Haussler 와의 협력 하에 학교에서 새로 구매한 50대의 컴퓨터를 리눅스 클러스터로 사용하여 한달 만에 이 시스템을 완성한다. Haussler와 협력이라고 하지만, 이 프로그램의 코드는 100% Kent 혼자서 한달 동안 밤낮없이 달려들어 완성한 것이었다. Kent가 완성한 이 프로그램을 이용해 비로소 다국적팀도 셀레라사에 뒤지지 않게 성공적으로 전체 인간 게놈 지도를 완성할 수 있었다. ( 정확히는 Kent의 프로그램을 이용한 다국적 팀이 3일 빨리 완성)
Kent가 생물학과 박사과정 학생이었지만, 사실 그는 3D graphics 엔진을 개발하는 컴퓨터 프로그래머로 일하던 전문 프로그래머였는데, 생물학에 매력을 느껴 community college에서 생물학 과목을 수강한 후, UCSC 생물학과 박사과정에 늦깍이 진학을 한 특이한 이력을 가지고 있던 학생이었다.
이 때의 성과를 바탕으로 UCSC는 UCSC genome browser라는 웹 기반 게놈 검색/분석 시스템을 서비스하게 되는데, 현재 이는 BLAST와 마찬가지로 생물학자들에게 매우 일상적으로 이용되는 Bioinformatics 서비스로 자리 메김했고, 덩달아 UCSC 는 그저그런 UC 계열 대학에서 세계 최정상의 게놈 연구 기관 중 한 곳으로 자리 메김 하게 되었다.
( Kent의 사례는 왜 다양한 background를 가진 직원/학생들을 창의적 업무를 요하는 회사/대학에서 뽑아야 하는지를 극명하게 보여주는 또하나의 좋은 예가 된다 )
Bioinformatics on the go
Bioinformatics 라는 분야는 Computational biology 라고 명명된 좀 더 수학적 이론에 치우친 분야를 토대로 게놈프로젝트 이전 1970년대 부터 이미 존재해 온 분야였지만, 인간게놈프로젝트를 기점으로 그 필요성과 역할이 크게 확대되었으며, 이에 대한 수요도 크게 늘어나게 되었다.
인간게놈프로젝트를 기점으로 비로소 독립된 Bioinformatics 교육 프로그램들이 미국의 대학들에 설립되기 시작했고( 연구 조직이 있었던 곳은 꾀나 있었지만, 독립적 학위 과정 프로그램을 가진 곳은 없었다), Michael Waterman이 1982년 부터 그 기틀을 잡아왔던 USC, 인간게놈프로젝트를 통해 유명해진 David Haussler 가 중심이 된 UCSC, 그외 Boston University 가 초기 독립된 Bioinformatics 학위 과정을 대학에 개설했는데, 이중 UCSC는 학부생 레벨( Bachelor degree)의 코스를 유일하게 설립해 운영을 시작했다.
국내에서는 2001년 숭실대학교에 최초의 학부 과정 학과가 개설(현재는 의생명공학부)되었고, 부산대학에 최초의 대학원 과정이 개설되었다. 2002년에는 서울대에 생물정보협동과정 대학원 과정이 개설되었고, 2003년에는 정문술 회장의 300억 기부로 KAIST에 Biosystems 이라는 이름으로 Bioinformatics 를 교육하는 학과(현재는 Bio & Brain Engineering 학과)가 만들어졌다.
인간게놈프로젝트에서 극명하게 드러난 것은 생물학이 이제 Large scale data 생산하는 시대에 접어들었고, 생물학이 이 large scale data의 분석을 통해 연구되는 Information science 즉 정보과학의 시대로 접어들었다는 사실이었다.
이는 게놈프로젝트 이후 생물학의 트렌드를 보면 극명히 드러나는데, 이 후에 생물학의 세부 분야들은 Genome 처럼 총체라는 뜻을 더해주는 -ome이 붙는 High-Thoughput data science 로 이름이 붙여졌고 ( Exressome, Proteome, Interactome, Metabolom , etc ) , 뛰어난 생물학 연구 팀에는 의례히 뛰어난 Bioinformatics 인력 혹은 공동연구팀이 함께 연구를 이끌어 가고 있음을 확인할 수 있다. 다시 말하면, 이제 첨단 생물학을 연구하기 위해서는 bioinformatics 로 대변되는 정보 과학적 분석 능력이 필수가 되었다는 말이 된다.
최근에 게놈 분석 비용이 획기적으로 낮아지면서, 게놈의 대중화와 이와 연계된 산업적 폭발이 목전에 와 있는데, 여기서도 게놈의 분석/관리/서비스를 위한 Bioinformatics 인력이 주도적 역할을 하게될 터인데,
게놈프로젝트 이후 많은 대학에 학위 과정이 개설되어 많은 인력을 배출해낸 미국에 비해 한국은 그나마 교육과정을 개설했던 학교들에서 오히려 Bioinformatics 이외의 분야들로 타겟을 바꾸어 학과를 포지셔닝 하면서, 제대로된 Bioinformatics 교육 커리큘럼을 갖춘 학교가 전무한 실정이다. 사실 미국을 제외하면 Bioinformatics 학위 과정이 활성화된 나라가 거의 없긴 한데, 그래서 자연히 앞으로의 Bioinformatics 분야도 미국이 주도적으로 발전시켜 나갈 수밖에 없지 않나 생각한다.
여담으로 현재 국내에서 중급 Bioinformatics 인력 조차도 구하기 쉽지 않은데, 앞으로 꾀나 오랜 시간 이런 흐름이 지속되지 않을가 조심스레 예측해 본다.
하지만, 1990년대 후반 크레이그 벤터( J. Craig Venter )가 셀레라(Celera genomics)사를 설립 대규모 자동 게놈 해독기를 장착하고 shortgun sequencing이라는 초고속 게놈 해독법을 이용해 인간 게놈을 분석하면서 이 지루한 프로젝트는 흥미진진한 소설 같은 상황으로 전개가 된다.
셀레라의 분석 기술로는 정부 주도의 다국적 팀에 의해 10년 이상이 소요될 것으로 예측되는 프로젝트를 불과 2-3년 안에 완료 할 수 있었고, 미생물 게놈을 빠른 시간 안에 분석 해 발표하면서 이는 기정 사실화 되었다. 정부 주도 하의 다국적 팀에게 이는 엄청난 위협이었고, 위기감을 불러 일으켰다. ( 과학자의 입장에서 보자면 수년간 노력한 일이 수포로 돌아가는 것과 마찬가지인데, 과학 연구에서 2등은 큰 의미가 없기 때문. 이와 관련한 스토리는 벤터의 자서전 Life decoded에 자세히 기록되어 있는데, 정부 주도 팀의 관료적 행태들은 구역질이 날 정도의 장면들이 많다. )
Shortgun genome sequencing
Shortgun sequencing 은 인간의 긴 30억개 염기 서열을 평균 500-1,000 개 염기 길이의 작은 단위로 잘라 읽은 후, 각각을 퍼즐 맞추기 식으로 이어 붙여 전체 30억개 염기 서열 부위를 완성하는 형태다.
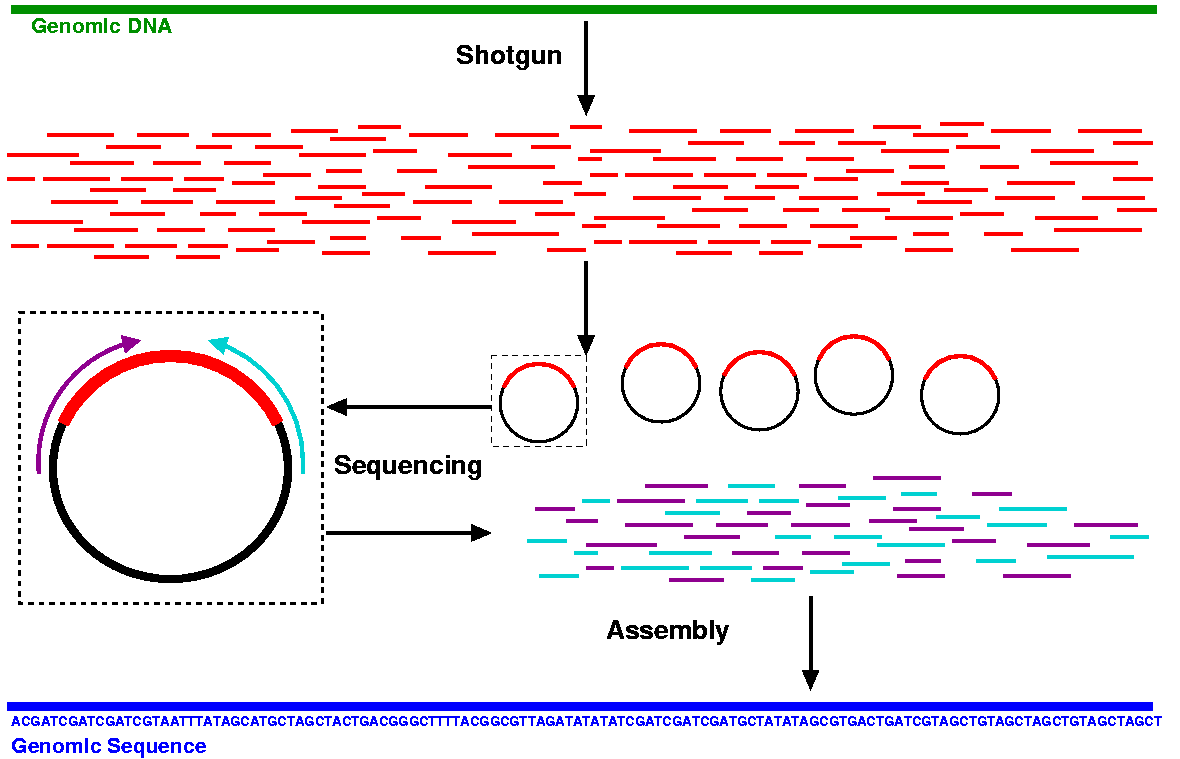 |
| Shortgun sequencing 방법 모식도 |
Shotgun sequencing 방법은 얼핏 매우 간단해 보이지만, 이 방법은 Bioinformatics 가 없이는 애초에 불가능한 서열 분석 방법이었다.
Shortgun sequencing을 통해 예를 들어 세개의 염기 서열 조각이 아래와 같이 주어진다고 하면 어떻게 이어 붙여야 할까?
AGTGT, GTAAC, ACTAG
아래와 같이 이어 붙이면 된다.
AGTGT
GTAAC
ACTAG
--------------------------
AGTGTAACTAG
아래와 같은 방식도 가능하다.
ACTAG
AGTGT
GTAAC
--------------------------
ACTAGTGTAAC
어떤 쪽이 옳은 것일까? 이런 서열 조각이 수천만개 있다면 어떨까? 어떻게 이 조각들을 제대로 꿰어 맞출 수 있을까? 이는 굉장히 정교한 수학적 모델을 필요로 했으며, 엄청난 컴퓨팅 파워를 필요로 하는 문제로 정통 생물학자들이 해결할 수 있는 문제가 아니었다.
이 문제를 푸는데 적용할 만한 수학적 이론은 Shortest common superstring problem (SSP), Eulerian graph problem 등이 있었고, Genome sequencing에 적용할 만한 이론들로 Michael Waterman 등의 기여로 대체로 정립,개발되어 왔지만, 실제로 이를 대량의 염기 서열 조각을 이어 붙이는 시스템에 적용하여 제작된 전례가 없었다.
생물정보학자 전쟁 :: Eugene Myers vs. Jim Kent
Eugene Myers 를 필두로 하는 Bioinformatics 팀이 셀레라 측의 Shortgun sequencing을 위한 염기 서열 조각 맞추기 문제를 위한 시스템을 성공적으로 완성했다. Gene Myers라고 흔히 불리는 Eugene Myers 는 NCBI에서 1990년 지금까지 가장 광범위하고 성공적으로 사용되는 생물학 프로그램 BLAST 를 공동 개발한 Bioinformatics 초기 영웅 중 한명이었는데, 그가 Celera에 합류하여 라떼 커피를 친구삼아 밤낮 가리지 않고 일에 몰두한 덕에 비소로 셀레라의 Shortgun sequencing 기술이 완성될 수 있었다.
Shortgun sequencing 시스템을 하드웨어적, 소프트웨어적으로 완벽히 갖추고 인간 게놈 분석의 막바지에 다다르고 있었던 셀레라 사에 비해, 다국적팀( 인간게놈컨소시움)은 정부의 중재로 셀레라 사와 함께 인간 게놈 분석 초안을 발표하기로 약속한 몇달 전이 되기 까지 이 게놈 조각 퍼즐 맞추기( Sequence assembly ) 를 위한 시스템이 완성되지 않아 초조하게 발을 구르는 상황이었다.
10년의 노력, 3조원의 정부 연구비를 정당화 하기 위해서 이 시스템은 너무나 간절했던 것이었다. 이 때, UCSC 생물학과에서 박사과정 학생이던 Jim Kent (William James Kent )가 David Haussler 와의 협력 하에 학교에서 새로 구매한 50대의 컴퓨터를 리눅스 클러스터로 사용하여 한달 만에 이 시스템을 완성한다. Haussler와 협력이라고 하지만, 이 프로그램의 코드는 100% Kent 혼자서 한달 동안 밤낮없이 달려들어 완성한 것이었다. Kent가 완성한 이 프로그램을 이용해 비로소 다국적팀도 셀레라사에 뒤지지 않게 성공적으로 전체 인간 게놈 지도를 완성할 수 있었다. ( 정확히는 Kent의 프로그램을 이용한 다국적 팀이 3일 빨리 완성)
Kent가 생물학과 박사과정 학생이었지만, 사실 그는 3D graphics 엔진을 개발하는 컴퓨터 프로그래머로 일하던 전문 프로그래머였는데, 생물학에 매력을 느껴 community college에서 생물학 과목을 수강한 후, UCSC 생물학과 박사과정에 늦깍이 진학을 한 특이한 이력을 가지고 있던 학생이었다.
이 때의 성과를 바탕으로 UCSC는 UCSC genome browser라는 웹 기반 게놈 검색/분석 시스템을 서비스하게 되는데, 현재 이는 BLAST와 마찬가지로 생물학자들에게 매우 일상적으로 이용되는 Bioinformatics 서비스로 자리 메김했고, 덩달아 UCSC 는 그저그런 UC 계열 대학에서 세계 최정상의 게놈 연구 기관 중 한 곳으로 자리 메김 하게 되었다.
( Kent의 사례는 왜 다양한 background를 가진 직원/학생들을 창의적 업무를 요하는 회사/대학에서 뽑아야 하는지를 극명하게 보여주는 또하나의 좋은 예가 된다 )
Bioinformatics on the go
Bioinformatics 라는 분야는 Computational biology 라고 명명된 좀 더 수학적 이론에 치우친 분야를 토대로 게놈프로젝트 이전 1970년대 부터 이미 존재해 온 분야였지만, 인간게놈프로젝트를 기점으로 그 필요성과 역할이 크게 확대되었으며, 이에 대한 수요도 크게 늘어나게 되었다.
인간게놈프로젝트를 기점으로 비로소 독립된 Bioinformatics 교육 프로그램들이 미국의 대학들에 설립되기 시작했고( 연구 조직이 있었던 곳은 꾀나 있었지만, 독립적 학위 과정 프로그램을 가진 곳은 없었다), Michael Waterman이 1982년 부터 그 기틀을 잡아왔던 USC, 인간게놈프로젝트를 통해 유명해진 David Haussler 가 중심이 된 UCSC, 그외 Boston University 가 초기 독립된 Bioinformatics 학위 과정을 대학에 개설했는데, 이중 UCSC는 학부생 레벨( Bachelor degree)의 코스를 유일하게 설립해 운영을 시작했다.
국내에서는 2001년 숭실대학교에 최초의 학부 과정 학과가 개설(현재는 의생명공학부)되었고, 부산대학에 최초의 대학원 과정이 개설되었다. 2002년에는 서울대에 생물정보협동과정 대학원 과정이 개설되었고, 2003년에는 정문술 회장의 300억 기부로 KAIST에 Biosystems 이라는 이름으로 Bioinformatics 를 교육하는 학과(현재는 Bio & Brain Engineering 학과)가 만들어졌다.
인간게놈프로젝트에서 극명하게 드러난 것은 생물학이 이제 Large scale data 생산하는 시대에 접어들었고, 생물학이 이 large scale data의 분석을 통해 연구되는 Information science 즉 정보과학의 시대로 접어들었다는 사실이었다.
이는 게놈프로젝트 이후 생물학의 트렌드를 보면 극명히 드러나는데, 이 후에 생물학의 세부 분야들은 Genome 처럼 총체라는 뜻을 더해주는 -ome이 붙는 High-Thoughput data science 로 이름이 붙여졌고 ( Exressome, Proteome, Interactome, Metabolom , etc ) , 뛰어난 생물학 연구 팀에는 의례히 뛰어난 Bioinformatics 인력 혹은 공동연구팀이 함께 연구를 이끌어 가고 있음을 확인할 수 있다. 다시 말하면, 이제 첨단 생물학을 연구하기 위해서는 bioinformatics 로 대변되는 정보 과학적 분석 능력이 필수가 되었다는 말이 된다.
최근에 게놈 분석 비용이 획기적으로 낮아지면서, 게놈의 대중화와 이와 연계된 산업적 폭발이 목전에 와 있는데, 여기서도 게놈의 분석/관리/서비스를 위한 Bioinformatics 인력이 주도적 역할을 하게될 터인데,
게놈프로젝트 이후 많은 대학에 학위 과정이 개설되어 많은 인력을 배출해낸 미국에 비해 한국은 그나마 교육과정을 개설했던 학교들에서 오히려 Bioinformatics 이외의 분야들로 타겟을 바꾸어 학과를 포지셔닝 하면서, 제대로된 Bioinformatics 교육 커리큘럼을 갖춘 학교가 전무한 실정이다. 사실 미국을 제외하면 Bioinformatics 학위 과정이 활성화된 나라가 거의 없긴 한데, 그래서 자연히 앞으로의 Bioinformatics 분야도 미국이 주도적으로 발전시켜 나갈 수밖에 없지 않나 생각한다.
여담으로 현재 국내에서 중급 Bioinformatics 인력 조차도 구하기 쉽지 않은데, 앞으로 꾀나 오랜 시간 이런 흐름이 지속되지 않을가 조심스레 예측해 본다.
Saturday, August 6, 2011
GigaScience :: BGI 가 출간하는 저널
BGI 가 BioMedCentral 과 합작하여 GigaScience라는 새로운 저널을 출간한다는 소식이다. ( http://www.gigasciencejournal.com/ )
'Big Data' 에 기반한 생의학 연구들의 출판을 목표로 하고 있고, 리뷰 과정에서 기존의 '학문적 의미'에 대한 잣대와 동시에 데이터의 유용성과 재연성을 중요한 잣대로 할 계획이라고 한다.
또하나 흥미로운 점은 '데이터셋' 자체에 DOI 를 부여하여, 데이터가 하나의 연구 논문에서 분석되는데서 벗어나 넓리 퍼져나가 다양한 연구에 이용될 수 있도록하여 데이터의 가치를 극대화하겠다는 점을 공언한 점이다.
"데이터의 '저장'과 함께 데이터의 효과적인 '전파' 도 중요한 문제다 "
- GigaScience 저널의 편집장 Scott Edmunds( BGI 소속)
데이터셋 자체에 DOI 를 부여하는 건 정말 '학계' 에서 보기 드문 획기적인 발상이다. 또한 이는 지난 10년 간의 Genomic data 들의 낮은 재사용성을 NGS data에 대해 답습하지 않을 수 있는 가능성을 어느 정도 열어 준다고 평가한다.
Microarray database인 NCBI 의 GEO에는 현재 24,000여개의 독립된 dataset (GSE 단위) 이 존재하지만, 두번 이상 재분석 된 dataset은 손에 꼽을 정도다. 기본적으로 어떤 논문에 어떤 GSE data가 쓰였는지 논문 본문을 검색하거나, 반대로 geo 데이터를 검색하지 않는 이상 알 수 없고, 해당 논문이 인용이 되어도, 논문에 딸린 dataset에 대한 인용인지를 평가하는 것도 어려운 등의 문제가 존재하여, dataset 자체에 대한 평가가 어려웠다는 문제가 있었기 때문이다.
1. Dataset 자체에 DOI 가 붙으면, 이를 분석한 논문과 별개로 dataset 자체에 대한 citation 이 가능해진다. 따라서 어떤 Dataset 이 퀄리티 높고, 재연성이 높은 dataset 인지를 일목요연하게 search하고 분석하는 것이 가능해 진다.
2. 인용이 가능해짐은 곧, dataset 자체로 높은 citation 을 얻을 수 있다는 얘기. 이는 곧, 높은 품질의 dataset 생산과 '전파' 에 대한 연구자들의 '동기'를 유발하게 하는 효과를 불러 일으킬 수 있다.
이는 다시 재연가능하고 유용한 dataset 숫자의 증가라는 긍정적인 결과로 이어진다.
2010년이 넘어가면서 본격적으로 생물학은 data dependent information science 로 넘어왔다. 바야흐로 bioinformatics 의 전성시대가 열리고 있고, dataset 자체가 논문과 상응하는 DOI 를 가질 수 있는 시대가 열렸다는 것은 이런 '시대의 흐름'을 보여주는 한 단면이 아닐까
'Big Data' 에 기반한 생의학 연구들의 출판을 목표로 하고 있고, 리뷰 과정에서 기존의 '학문적 의미'에 대한 잣대와 동시에 데이터의 유용성과 재연성을 중요한 잣대로 할 계획이라고 한다.
또하나 흥미로운 점은 '데이터셋' 자체에 DOI 를 부여하여, 데이터가 하나의 연구 논문에서 분석되는데서 벗어나 넓리 퍼져나가 다양한 연구에 이용될 수 있도록하여 데이터의 가치를 극대화하겠다는 점을 공언한 점이다.
"데이터의 '저장'과 함께 데이터의 효과적인 '전파' 도 중요한 문제다 "
- GigaScience 저널의 편집장 Scott Edmunds( BGI 소속)
데이터셋 자체에 DOI 를 부여하는 건 정말 '학계' 에서 보기 드문 획기적인 발상이다. 또한 이는 지난 10년 간의 Genomic data 들의 낮은 재사용성을 NGS data에 대해 답습하지 않을 수 있는 가능성을 어느 정도 열어 준다고 평가한다.
Microarray database인 NCBI 의 GEO에는 현재 24,000여개의 독립된 dataset (GSE 단위) 이 존재하지만, 두번 이상 재분석 된 dataset은 손에 꼽을 정도다. 기본적으로 어떤 논문에 어떤 GSE data가 쓰였는지 논문 본문을 검색하거나, 반대로 geo 데이터를 검색하지 않는 이상 알 수 없고, 해당 논문이 인용이 되어도, 논문에 딸린 dataset에 대한 인용인지를 평가하는 것도 어려운 등의 문제가 존재하여, dataset 자체에 대한 평가가 어려웠다는 문제가 있었기 때문이다.
1. Dataset 자체에 DOI 가 붙으면, 이를 분석한 논문과 별개로 dataset 자체에 대한 citation 이 가능해진다. 따라서 어떤 Dataset 이 퀄리티 높고, 재연성이 높은 dataset 인지를 일목요연하게 search하고 분석하는 것이 가능해 진다.
2. 인용이 가능해짐은 곧, dataset 자체로 높은 citation 을 얻을 수 있다는 얘기. 이는 곧, 높은 품질의 dataset 생산과 '전파' 에 대한 연구자들의 '동기'를 유발하게 하는 효과를 불러 일으킬 수 있다.
이는 다시 재연가능하고 유용한 dataset 숫자의 증가라는 긍정적인 결과로 이어진다.
2010년이 넘어가면서 본격적으로 생물학은 data dependent information science 로 넘어왔다. 바야흐로 bioinformatics 의 전성시대가 열리고 있고, dataset 자체가 논문과 상응하는 DOI 를 가질 수 있는 시대가 열렸다는 것은 이런 '시대의 흐름'을 보여주는 한 단면이 아닐까
Monday, June 27, 2011
iPAD APP:: Bio-IT World
아이패드는 기존의 컨텐츠를 완전히 다른 레벨의 컨텐츠로 진화를 가능케 해주는 매우 뛰어난, 새로운 영역의 기계다. (적어도 내게는)PC에서의 꾸리한 인터페이스에 거의 사용도 안 하던 youtube가 가장 우수한 동영상 컨텐츠 뷰어가 되고, 웹툰이라는 컨텐츠가 '다음 코믹스' 아이패드 앱을 통해 내게는 완전히 새로운 형태의 만화 컨텐츠로 격상되는 경험을 주었다.
BioITWorld 아이패드 앱도 역시 이처럼 기존의 컨텐츠가 완전히 새로운 경험을 담은 우수한 컨텐츠로 새롭게 다가오게 만든 앱이다. BioITWorld 는 생물학과 컴퓨터 공학의 접점, 즉 Bioinformatics 라 명명하는 영역의 다양한 뉴스와 깊이 있는 분석 보고서를 담아 2달에 한번씩 발간되는 잡지다. 무료로 제공되기 때문에 주로 웹을 통해서 볼 수밖에 없는데, 웹을 통해 발간된 잡지는 오랜 시간 컴퓨터 모니터를 통해 읽기가 힘들어 제대로 컨텐츠 소비를 하기 어려운 단점이 있었다.
그러던 것이 이번 BioITWorld 아이패드 앱을 통해, 종이 잡지 보다도 훨씬 읽기 편한, interactiive 한 컨텐츠를 효과적으로 전달하는 잡지로 거듭났다고 개인적으로 평가해본다.
앱을 실행시키면 위와 같이 Archive 화면이 뜨고, 원하는 달의 잡지를 볼 수 있다.


기사를 클릭하면 왼쪽과 같이 기사를 보여주는데, 이 상태론 글씨가 너무 작아서 제대로 읽을 수가 없다. 이제껏 웹 버전( 및 이를 출력한 프린트 버전 ) 은 이런 화면에서 잡지를 읽어야 했기 때문에, 제대로 내용을 읽기가 힘들고, 설령 읽는다해도 PC 화면을 붙잡고 오래 읽어내기는 힘들었다. 이제 아이패드용 앱에서 오른쪽과 같이 자유자재로 확대해서 편하게 내용을 읽을 수 있다.
웹버전 잡지 답게, 내용에 맞는 동영상이 링크되어 있고, 이를 누르면 바로 Youtube에 등록된 동영상을 볼 수 있다.
BioIT world 아이패드 앱으로 Bioinformatics 뉴스를 실감나게 즐겨보자!
BioITWorld 아이패드 앱도 역시 이처럼 기존의 컨텐츠가 완전히 새로운 경험을 담은 우수한 컨텐츠로 새롭게 다가오게 만든 앱이다. BioITWorld 는 생물학과 컴퓨터 공학의 접점, 즉 Bioinformatics 라 명명하는 영역의 다양한 뉴스와 깊이 있는 분석 보고서를 담아 2달에 한번씩 발간되는 잡지다. 무료로 제공되기 때문에 주로 웹을 통해서 볼 수밖에 없는데, 웹을 통해 발간된 잡지는 오랜 시간 컴퓨터 모니터를 통해 읽기가 힘들어 제대로 컨텐츠 소비를 하기 어려운 단점이 있었다.
그러던 것이 이번 BioITWorld 아이패드 앱을 통해, 종이 잡지 보다도 훨씬 읽기 편한, interactiive 한 컨텐츠를 효과적으로 전달하는 잡지로 거듭났다고 개인적으로 평가해본다.
 |
| BioIT World 아이패드 앱, 초기 Archive 화면 |


기사를 클릭하면 왼쪽과 같이 기사를 보여주는데, 이 상태론 글씨가 너무 작아서 제대로 읽을 수가 없다. 이제껏 웹 버전( 및 이를 출력한 프린트 버전 ) 은 이런 화면에서 잡지를 읽어야 했기 때문에, 제대로 내용을 읽기가 힘들고, 설령 읽는다해도 PC 화면을 붙잡고 오래 읽어내기는 힘들었다. 이제 아이패드용 앱에서 오른쪽과 같이 자유자재로 확대해서 편하게 내용을 읽을 수 있다.
 |
| 동영상이 삽입된 페이지 ( 붉은색 카메라 아이콘) |
 |
| 위 페이지에서 동영상 아이콘을 누르면 연결되는 페이지 |
웹버전 잡지 답게, 내용에 맞는 동영상이 링크되어 있고, 이를 누르면 바로 Youtube에 등록된 동영상을 볼 수 있다.
BioIT world 아이패드 앱으로 Bioinformatics 뉴스를 실감나게 즐겨보자!
Sunday, February 6, 2011
생명정보학자들은 어떤 역할을 해야 하나?
네이쳐 바이오테크놀로지에 2010년 생명정보학 연구 하일라이트에 관한 글이 실렸다.
http://www.nature.com/nbt/journal/v29/n1/pdf/nbt.1747.pdf
이 글 중 Box2 에는 '분야를 뛰어넘는 연구자( Cross functional individual )' 들이 생명정보학 연구에 어떤 기여를 하고 있는지에 관한 내용이 담겨있다. 여기서 말하는 분야를 뛰어넘는 연구자들은 곧, 생명정보학자, 계산 생물학자로 지칭되는 생물학과 계산과학 두 분야 모두에 전문적인 지식을 가진 연구자들을 이야기 한다.
생명정보학 툴이 생물학계 전반에 퍼지는 양상은 3단계에 거쳐 진행이 된다고 하는데, 이 순서는 아래와 같다.
1. 생명정보학자들이 정보학적 분석을 통해 해결할 수 있는 생물학 문제들을 인식하고 간단한 방법론을 통해 이런 문제들을 해결할 수 있다는 사실을 증명한다.
2. 정통 계산과학자 ( 수학 및 컴퓨터과학자들 ) 들이 좀더 정교한 방법론들을 이용해 생명정보학자들이 만들어 놓은 간단한 방법론을 개선하여 생물학자들이 사용하기 쉬운 '툴'로 만든다.
3. 만들어진 '툴'을 필요로 하는 생물학자들이 사용하여 새로운 발견에 이용된다.
마이크로어레이 '분류( classification )' 연구에서 핵심이 되는 것은 특징 찾기(feature selection) 과정인데, 이 분야 연구를 예로 들어보면, 1999년 Todd golub 에 의해 출판된 네이쳐 논문이 선구적인 논문으로 이와 관련해 가장 인용이 많이 된 논문인 것으로 알고 있다.
그런데, 이 논문의 핵심이 되는 feature selection 알고리즘은 새롭게 개발된 것이 아니라, 흔하게 쓰이는 t-test 다. 즉, 위의 1번 과정 처럼 '기존에 존재하는 단순한 방법론' 을 '새로운 생물학 문제'에 적용을 하여 효과적으로 문제 해결을 할 수 있다는 사실을 증명한 것이다.
이후로 이 보다 훨씬 정교한 방법론들이 수학자, 통계학자, 컴퓨터 공학자들에 의해 엄청나게
만들어 지면서 2번 과정이 진행된다. 이 중에 가장 효과적이라고 검증된 SAM 과 같은 몇몇개의 방법론들은 마이크로어레이 분류 문제 해결을 위해 전세계의 생물학자및 생명정보학 연구자들에 의해 사용되는 3번 과정을 거치며 기술 정착 단계에 이른다.
1,2,3번 과정에서 가장 큰 업적으로 평가받는 것은 1번, 즉 가장 먼저 '문제를 인식' 하고 '간단한 방법으로 해결 가능성' 을 보인 사람이다. 노벨상도 이런 일을 한 사람에게 돌아간다. 즉, 노벨상이나 각광받는 연구 업적들은 대단한 '방법론' 에 의한 경우보다 새로운 '문제 인식(발견)' 인 경우가 많고, 이런 새로운 '문제 인식' 에는 복잡한 방법론이 필요하지 않은 경우가 많다는 얘기다.
똑같은 문제를 조금 더 효과적으로 해결하는 방법론 을 만들기 위해 노력하는 것 보다, '중요한 문제' 발굴에 좀 더 신경을 쓰는 것이 좋은 생명정보학 연구자로 거듭나는 길이 되지 않을까 생각해 본다.
http://www.nature.com/nbt/journal/v29/n1/pdf/nbt.1747.pdf
이 글 중 Box2 에는 '분야를 뛰어넘는 연구자( Cross functional individual )' 들이 생명정보학 연구에 어떤 기여를 하고 있는지에 관한 내용이 담겨있다. 여기서 말하는 분야를 뛰어넘는 연구자들은 곧, 생명정보학자, 계산 생물학자로 지칭되는 생물학과 계산과학 두 분야 모두에 전문적인 지식을 가진 연구자들을 이야기 한다.
생명정보학 툴이 생물학계 전반에 퍼지는 양상은 3단계에 거쳐 진행이 된다고 하는데, 이 순서는 아래와 같다.
1. 생명정보학자들이 정보학적 분석을 통해 해결할 수 있는 생물학 문제들을 인식하고 간단한 방법론을 통해 이런 문제들을 해결할 수 있다는 사실을 증명한다.
2. 정통 계산과학자 ( 수학 및 컴퓨터과학자들 ) 들이 좀더 정교한 방법론들을 이용해 생명정보학자들이 만들어 놓은 간단한 방법론을 개선하여 생물학자들이 사용하기 쉬운 '툴'로 만든다.
3. 만들어진 '툴'을 필요로 하는 생물학자들이 사용하여 새로운 발견에 이용된다.
마이크로어레이 '분류( classification )' 연구에서 핵심이 되는 것은 특징 찾기(feature selection) 과정인데, 이 분야 연구를 예로 들어보면, 1999년 Todd golub 에 의해 출판된 네이쳐 논문이 선구적인 논문으로 이와 관련해 가장 인용이 많이 된 논문인 것으로 알고 있다.
그런데, 이 논문의 핵심이 되는 feature selection 알고리즘은 새롭게 개발된 것이 아니라, 흔하게 쓰이는 t-test 다. 즉, 위의 1번 과정 처럼 '기존에 존재하는 단순한 방법론' 을 '새로운 생물학 문제'에 적용을 하여 효과적으로 문제 해결을 할 수 있다는 사실을 증명한 것이다.
이후로 이 보다 훨씬 정교한 방법론들이 수학자, 통계학자, 컴퓨터 공학자들에 의해 엄청나게
만들어 지면서 2번 과정이 진행된다. 이 중에 가장 효과적이라고 검증된 SAM 과 같은 몇몇개의 방법론들은 마이크로어레이 분류 문제 해결을 위해 전세계의 생물학자및 생명정보학 연구자들에 의해 사용되는 3번 과정을 거치며 기술 정착 단계에 이른다.
1,2,3번 과정에서 가장 큰 업적으로 평가받는 것은 1번, 즉 가장 먼저 '문제를 인식' 하고 '간단한 방법으로 해결 가능성' 을 보인 사람이다. 노벨상도 이런 일을 한 사람에게 돌아간다. 즉, 노벨상이나 각광받는 연구 업적들은 대단한 '방법론' 에 의한 경우보다 새로운 '문제 인식(발견)' 인 경우가 많고, 이런 새로운 '문제 인식' 에는 복잡한 방법론이 필요하지 않은 경우가 많다는 얘기다.
똑같은 문제를 조금 더 효과적으로 해결하는 방법론 을 만들기 위해 노력하는 것 보다, '중요한 문제' 발굴에 좀 더 신경을 쓰는 것이 좋은 생명정보학 연구자로 거듭나는 길이 되지 않을까 생각해 본다.
Sunday, April 25, 2010
University ranking on Bioinformatics [ 생명정보학 대학 랭킹 ]
METHOD
- Search journal : I used CPAN perl module 'WWW::Search::PubMed' to retrieve information of papers published in three journals. To retieve Affiliation information, I added lines of codes.
- Control variation of university name : Majority of papers used standard official name to specify each university. Howevr, some of papers used university name which is subtly different from the standard one. Since it requires mandatory validation, I didn't consider these variations. I only consider universities are the same if they used exactly the same name. However, I controled terms for 'University of California' . There are branches of universities for 'University of California', such as UC san diego, UC los angeles , UC berkely, etc. And generally, the university name is divided by comma (, ) which is a seperator in affiliation line into two parts, 'University of California' and city name such as 'Los Angeles', 'San diego' , etc. So I combined university name and the city name if 'University of California' is stated alone followed by comma without city name.
Overall ranking ( Top 50 )
#colum 1: Ranking
#colum 2: Number of publications
#colum 3: University name
( Non US universities are orange colored )
1 108 UNIVERSITY OF CALIFORNIA SAN DIEGO
2 88 STANFORD UNIVERSITY
3 72 COLUMBIA UNIVERSITY
3 72 UNIVERSITY OF WASHINGTON
3 72 UNIVERSITY OF MANCHESTER
6 71 UNIVERSITY OF MICHIGAN
7 69 YALE UNIVERSITY
8 63 UNIVERSITY OF CALIFORNIA BERKELEY
8 63 UNIVERSITY OF CAMBRIDGE
10 60 WASHINGTON UNIVERSITY
11 59 UNIVERSITY OF OXFORD
12 58 TEL AVIV UNIVERSITY
12 58 UNIVERSITY OF TOKYO
14 56 NATIONAL UNIVERSITY OF SINGAPORE
15 53 PRINCETON UNIVERSITY
16 52 UNIVERSITY COLLEGE LONDON
17 48 UNIVERSITY OF MINNESOTA
17 48 UNIVERSITY OF QUEENSLAND
19 47 IOWA STATE UNIVERSITY
20 45 BOSTON UNIVERSITY
21 44 UNIVERSITY OF TORONTO
22 42 UNIVERSITY OF CALIFORNIA IRVINE
22 42 UNIVERSITY OF PITTSBURGH
24 41 CORNELL UNIVERSITY
24 41 KYOTO UNIVERSITY
24 41 UNIVERSITY OF SOUTHERN CALIFORNIA
27 40 UNIVERSITY OF CALIFORNIA SAN FRANCISCO
28 39 KAIST ( Korea Advanced Institute of Science & Technology )
28 39 TEXAS A&M UNIVERSITY
28 39 UNIVERSITY OF BRITISH COLUMBIA
31 38 JOHNS HOPKINS UNIVERSITY
32 37 HARVARD UNIVERSITY
32 37 UNIVERSITY OF CALIFORNIA DAVIS
34 36 UNIVERSITY OF PENNSYLVANIA
35 35 CARNEGIE MELLON UNIVERSITY
35 35 UPPSALA UNIVERSITY
35 35 UNIVERSITY OF MARYLAND
38 33 BIELEFELD UNIVERSITY
39 31 UNIVERSITY OF CALIFORNIA SANTA CRUZ
40 30 PEKING UNIVERSITY
40 30 UNIVERSITY OF TEXAS SOUTHWESTERN MEDICAL CENTER
40 30 UNIVERSITY OF GEORGIA
43 29 INDIANA UNIVERSITY
43 29 UNIVERSITY OF GLASGOW
43 29 UNIVERSITY OF CALIFORNIA LOS ANGELES
43 29 TSINGHUA UNIVERSITY
47 28 OHIO STATE UNIVERSITY
48 27 Seoul National University
49 26 TECHNICAL UNIVERSITY OF DENMARK
49 26 PENNSYLVANIA STATE UNIVERSITY
Rising universities
After HGP, bioinformatics recogenized as one of the most important field of genome revolution. Universities that did not specially drive their effort on bioinformatics before the time had started bioinformatics research with large investment. Therfore, the overall ranking might have undergone lots of changes.
To find these variation, I ranked again according to relative rank increase( rank_after_2001 - rank_before_2005 / rank_before_2001).
Range of publication time is divided into two duration, before 2001 and after 2005.
Universities without any publication before 2001 were not considered for this analysis.
[ Top 10 raising university]
#colum 1: Ranking
#colum 2: University name
#colum 3: relative rank difference
#colum 4: ranking for 1998-2000
#colum 5: ranking for 2006-2010
1.UNIVERSITY OF CALIFORNIA SAN DIEGO 0.93 15 1
2.UNIVERSITY OF WASHINGTON 0.92 39 3
3.STANFORD UNIVERSITY 0.66 6 2
4.TEL AVIV UNIVERSITY 0.56 39 17
4.UNIVERSITY OF QUEENSLAND 0.56 39 17
6.YALE UNIVERSITY 0.5 6 3
7.IOWA STATE UNIVERSITY 0.41025641025641 39 23
8.UNIVERSITY OF SOUTHERN CALIFORNIA 0.35 39 25
8.UNIVERSITY OF TORONTO 0.35 39 25
8.KYOTO UNIVERSITY 0.35 39 25
Thoughts on the ranking
1. Can I say that high ranked universities are better than universities in low ranked?
- University is different from research institution. I think university ranking should consider not only research performance, but also how successfully they educate student. This ranking is based only on number of publications so that it only consider research performance. Therefore, this ranking should not be considered to reveal overall value of universities.
2. How does UCSD excel the other top schools?
- UCSD is ranked around 7-10 when the ranking is based only on 'Bioinformatics' or 'BMC bioinformatics'. However, UCSD is top ranked with overwelming number of publications when it comes to 'Plos computational biology' journal. Since Plos family journals were initiated with tight relationship with faculty members at UCSD, they have willingness to publish papers in Plos journal. Moreover, Philip Bourne, a professor at UCSD in the field of bioinformatics, has been contributed a series of editorials, ' Ten simple rule ' advise series, in Plos Computational biology, which are not research papers, but counted as equally as research papers to be considered for ranking.
- As I described in the beginning, this ranking is only based on top three journals in the field of bioinformatics or computatinoal biology. If the other journals were considered, the ranking would be changed a lot.
- In the top 10 list, only Cambridge and Manchester are universities out of USA. In top 20, there are 8 and 12 in top 30. Regardless of drawbacks of ranking criteria, it's ture that USA is a leading country in this field.
- List of countries other than USA in top 30 ranking are Japan, Canada, Israel, Korea, Singapore and Austrailia. 3 Aisan countries have 4 universities ranked within top 30.
Tuesday, March 30, 2010
시퀀스에서 Di-nucleotide frequency 계산 문제.
지난해 pung96님의 블로그에 올라왔던 Di-nucleotide composition frequency 세기 문제( http://perlog.pung96.net/20 ) 를 다시 한번 언급해 본다.
문제를 다시 정리해 보면,
AGATAGCGATAGCG
AGATGACGATAGAG
...
위 처럼 DNA 서열이 담긴 input 파일이 있을 때, DNA base 두개씩을 끊은 dinucleotide ( e.g., AG, CT ) 와 같은 16개 조합( DNA base가 4개 이므로 2개의 조합 가지 수는 16개 )의 빈도를 계산하는 것이 원글의 문제다. 이때, 앞에서 순서대로 2개씩 읽되, window size는 1로 하여 1칸씩 옮겨가며 2개씩 읽어 조합을 계산해야 한다.
구체적으로 AGAA 네개 DNA 서열인 경우, AG, GA ,AA 세개의 dinucleotide 조합으로 계산해야 한다는 것이다.
TIMTOWTDI, perl의 특성답게 이를 해결하는 방법은 상당히 다양한데, pung96님의 글에 언급되지 않은 내용을 추가로 정리해 보고자 한다.
substr을 이용하는 경우 ( DNA 서열은 $seq에 저장되어 있는 경우 )
정규식의 lookahead 이용하는 경우,
이걸 한줄로 줄이면,
반복을 while로 돌리면,
map 에 넣으면,
와 같은 방식으로도 문제를 해결할 수 있다. 포스팅의 요지는 lookahead 를 이용한 정규식 사용!!
사족을 좀 덧붙여 보면, 이 문제는 연세대 생명공학과 대학원 이인석 교수의 bioinformatics 수업 과제 문제다. 지난해와 올해 연구소에 연대 대학원 학생분들이 있어, 내게 이 문제에 대한 조언을 구해와 본의 아니게 2년 연속으로 이 문제에 대해 생각해볼 기회가 생겨, 포스팅해 본다.
문제를 다시 정리해 보면,
AGATAGCGATAGCG
AGATGACGATAGAG
...
위 처럼 DNA 서열이 담긴 input 파일이 있을 때, DNA base 두개씩을 끊은 dinucleotide ( e.g., AG, CT ) 와 같은 16개 조합( DNA base가 4개 이므로 2개의 조합 가지 수는 16개 )의 빈도를 계산하는 것이 원글의 문제다. 이때, 앞에서 순서대로 2개씩 읽되, window size는 1로 하여 1칸씩 옮겨가며 2개씩 읽어 조합을 계산해야 한다.
구체적으로 AGAA 네개 DNA 서열인 경우, AG, GA ,AA 세개의 dinucleotide 조합으로 계산해야 한다는 것이다.
TIMTOWTDI, perl의 특성답게 이를 해결하는 방법은 상당히 다양한데, pung96님의 글에 언급되지 않은 내용을 추가로 정리해 보고자 한다.
substr을 이용하는 경우 ( DNA 서열은 $seq에 저장되어 있는 경우 )
for( my $i=0; $i< length( $seq ) - 1 ;$i++ ){
$count{ substr($seq,$i,2) }++;
}
정규식의 lookahead 이용하는 경우,
my @di=$seq=~/(?=(\w{2}))/g;
$count{$_}++ for @di;
이걸 한줄로 줄이면,
$count{$_}++ for $seq=~/(?=(\w{2}))/g;
반복을 while로 돌리면,
$count{$1}++ while $seq=~/(?=(\w{2}))/g;
map 에 넣으면,
map{ $count{$_}++ } $seq=~/(?=(\w{2}))/g;
와 같은 방식으로도 문제를 해결할 수 있다. 포스팅의 요지는 lookahead 를 이용한 정규식 사용!!
사족을 좀 덧붙여 보면, 이 문제는 연세대 생명공학과 대학원 이인석 교수의 bioinformatics 수업 과제 문제다. 지난해와 올해 연구소에 연대 대학원 학생분들이 있어, 내게 이 문제에 대한 조언을 구해와 본의 아니게 2년 연속으로 이 문제에 대해 생각해볼 기회가 생겨, 포스팅해 본다.
Thursday, February 11, 2010
Dynamic modularity for disease prediction
2009년 2월에 출간된 Dynamic modularity in protein interaction networks predicts breast cancer outcome ( nature biotech, 27, 199 ) 에서 'Dynamic modularity' 라는 개념을 토대로 PPI network 을 분석하고, breast cancer 환자 survival data를 이용 환자 survival을 prediction 한 연구 내용을 소개하고 있다.
기본적으로 Dynamic modularity라는 개념은 이미 이전에 많이 언급되었던 내용인데, network의 연결이 상황에 따라 느슨하거나 타이트하게 변화한다는 것이 핵심이 된다.
논문에서는 우선 분석 대상이 되는 유전자를 network의 허브 유전자로 한정한다. 이렇게 허브 유전자로 한정한 원인은 두가지 정도 생각해 볼 수 있는데, 첫째, network 의 허브들 간의 연결에 주목하여 중요한 biological path 를 커버하여 분석할 수 있고, 동시에 주요하지 않은 유전자들을 모두 분석에 사용하여 과다하게 복잡한 양의 결과 분석을 통해 올 수 있는 노이즈를 사전에 제거할 수 있다는 의미, 두번째로는 분석 계산양의 감소 효과다. 논문에서는 허브 유전자간 co-expression 계산을 통해 네트워크의 dynamic modularity 를 잡아내는데, 네트웍에 들어있는 모든 유전자들을 사용한 pair-wise co-expression 계산량은 node 개수가 증가함에 따라 기하급수적으로 늘어나 계산 시간을 엄청나게 늘린다. 40,000개 노드 정도가 되면 array 숫자에 따라 틀리긴 하겠으나 경험상 일주일 이상 하나의 컴퓨터가 온전하게 소요되어야 할 정도 ( 포스팅 참조 ).
한정된 hub 유전자들을 두고, tissue-specific human gene expression dataset을 이용해 1)intramodular hub 와 2)intermodular hub 로 허브 유전자들을 구분한다. 이때, intramodular hub 는 tissue non-specific하게 높은 co-expression 정도를 보여주는 유전자들이고, intermodular는 tissue에 따른 co-expression 이 변화량이 들쭉날쭉한 유전자들이 된다.
논문에서는 이렇게 구분된 두개 클래스의 허브 유전자들에 대한 network topology, functional analysis 등을 통해 intra-,inter-modular hub 유전자들의 특성을 비교 분석한다.
마지막으로 breast cancer patient cohort data를 이용해 breast cancer patient 에 specific하게 강하거나 약한 co-expression 을 보이는 허브 유전자 쌍들을 feature로 이용해 prediction 하고 그 결과를 리포팅하고 있는데, 결과는 현재 상업적으로 이용되는 breast cancer diagnostics 기법에 비해 6~23% 높은 정확도를 보인다.
기본적으로 Dynamic modularity라는 개념은 이미 이전에 많이 언급되었던 내용인데, network의 연결이 상황에 따라 느슨하거나 타이트하게 변화한다는 것이 핵심이 된다.
논문에서는 우선 분석 대상이 되는 유전자를 network의 허브 유전자로 한정한다. 이렇게 허브 유전자로 한정한 원인은 두가지 정도 생각해 볼 수 있는데, 첫째, network 의 허브들 간의 연결에 주목하여 중요한 biological path 를 커버하여 분석할 수 있고, 동시에 주요하지 않은 유전자들을 모두 분석에 사용하여 과다하게 복잡한 양의 결과 분석을 통해 올 수 있는 노이즈를 사전에 제거할 수 있다는 의미, 두번째로는 분석 계산양의 감소 효과다. 논문에서는 허브 유전자간 co-expression 계산을 통해 네트워크의 dynamic modularity 를 잡아내는데, 네트웍에 들어있는 모든 유전자들을 사용한 pair-wise co-expression 계산량은 node 개수가 증가함에 따라 기하급수적으로 늘어나 계산 시간을 엄청나게 늘린다. 40,000개 노드 정도가 되면 array 숫자에 따라 틀리긴 하겠으나 경험상 일주일 이상 하나의 컴퓨터가 온전하게 소요되어야 할 정도 ( 포스팅 참조 ).
한정된 hub 유전자들을 두고, tissue-specific human gene expression dataset을 이용해 1)intramodular hub 와 2)intermodular hub 로 허브 유전자들을 구분한다. 이때, intramodular hub 는 tissue non-specific하게 높은 co-expression 정도를 보여주는 유전자들이고, intermodular는 tissue에 따른 co-expression 이 변화량이 들쭉날쭉한 유전자들이 된다.
논문에서는 이렇게 구분된 두개 클래스의 허브 유전자들에 대한 network topology, functional analysis 등을 통해 intra-,inter-modular hub 유전자들의 특성을 비교 분석한다.
마지막으로 breast cancer patient cohort data를 이용해 breast cancer patient 에 specific하게 강하거나 약한 co-expression 을 보이는 허브 유전자 쌍들을 feature로 이용해 prediction 하고 그 결과를 리포팅하고 있는데, 결과는 현재 상업적으로 이용되는 breast cancer diagnostics 기법에 비해 6~23% 높은 정확도를 보인다.
Tuesday, January 19, 2010
Median Rank Score ( MRS ) for cross-platform microarray classification
Patrick Warnat의 논문( BMC bioinformatics, 6, 265 ) 을 읽던 중, Median Rank Score 을 이용한 cross-platform microarray data integration 방법이 있어 정리해 본다.
[Procedure]
Procedure를 보면 quantile normalization 방법의 기본적인 아이디어와 유사하다는 것을 알 수 있는데, 자연스럽게 MRS 를 이용한 integration 결과도 quantile normalization과 유사하게 서로 다른 range의 expression value를 공통된 range의 expression value로 normalization 하게 된다.
[Procedure]
- 서로 다른 Microarray dataset 두개를 통합하는 경우, n개 array 로 이루어진 datasetA 와 m개 array로 이루어진 datasetB 가 있다고 가정하고, 먼저 둘 중 하나의 dataset을 reference dataset으로 정한다.
- Reference dataset의 각 유전자( probeset) 에 대해 median expression value 를 구한다
- 유전자에 대한 Median expression value vector를 오름차순으로 sort
- Non-reference dataset의 각 array를 expression value를 기준으로 오름차순으로 sort
- Sort된 non-reference dataset의 array 의 각 유전자의 expression value를 같은 순위의 reference dataset의 median expression value vector의 값으로 대치한다.
Procedure를 보면 quantile normalization 방법의 기본적인 아이디어와 유사하다는 것을 알 수 있는데, 자연스럽게 MRS 를 이용한 integration 결과도 quantile normalization과 유사하게 서로 다른 range의 expression value를 공통된 range의 expression value로 normalization 하게 된다.
Wednesday, January 13, 2010
생명정보학( Bioinformatics, Computational biology) 선도 연구기관 랭킹
1000 genome project ( 1000명의 genome을 sequencing 하는 HGP 이후의 가장 큰 국제 생물학 연구 컨소시움 ) 에 참여하는 연구 그룹 중, 실험 부분을 제외한 순수 data analysis 참여자들의 숫자를 counting 하고 이 순서로 생명정보학(Bioinformatics, Computational biology) 대학/연구그룹 랭킹을 매겨봤다.
HGP, 1000 genome project 를 리딩하는 sanger 연구소가 1위, MIT와 하바드의 broad institute이 2위로 bioinformaitcs 분야를 이끌고 있는걸 확인할 수 있다.
미국 탑 대학들 중 칼텍이나 스탠포드는 분석 참여자 0명과 1명으로 이 순위 최하위권에 랭크되어 있다. 시퀀싱 프로젝트인 만큼 업계 리더들인 Applied biosystems 와 Illumina 가 10위 12위를 마크하고 있다. 또 아시아권에선 유일하게 Beijing Genome Institute이 미국의 대표적인 Bioinformatics 기관인 NCBI, Illumina와 공동으로 12위를 마크하고 있다. 유럽의 연구기관들은 1위를 차지한 Sanger institute, 5위의 Oxford, 7위 EBI, 17위의 MPI ( Max Planck Institute ) 이 상위권에 랭크되었다.
향후, Personalized genomics 가 생물학 연구의 핵심화두이자, 바이오 산업 중흥의 열쇠를 쥐고 있다는 것을 생각하면, 앞으로 박사과정 및 포닥을 위한 연구기관 선정을 고려하고 있는 경우, 이 리스트에 포함된 연구기관들과 연구자들을 적극 고려해 봄직 하다고 생각한다.
#### 1,000 Genome project, Data analysis 참여 연구자 숫자에 따른 생명정보학 연구기관 랭킹 ####
1. Sanger Institute 30
2. Broad Institute 26
3. Yale University 14
4. Cornell University 13
5. Oxford University 11
6. Baylor College of Medicine 10
6. European Bioinformatics Institute 10
6. Washington University in St. Louis 10
9. University of Chicago 9
10. Applied Biosystems 7
10. University of Michigan 7
12. Illumina 6
12. Beijing Genomics Institute, Shenzhen 6
12. National Center for Biotechnology Information 6
15. Boston College 5
15. University of California, Santa Cruz 5
17. Cold Spring Harbor Laboratory 4
17. University of Washington 4
17. Max Planck Institute for Molecular Genetics 4
20. Roche Applied Science 3
20. Louisiana State University 3
22. University of Utah 2
22. Brigham and Women's Hospital / Broad / Harvard 2
22. University of Auckland 2
22. National Human Genome Research Institute 2
22. Translational Genomic Research Institute 2
22. University of California, San Diego 2
Monday, December 7, 2009
유전자 발현 정보 이용한 Pathway based classification
Microarray gene expression data를 이용한 disease classifier 연구는 microarray 연구 초창기 부터 많은 연구자들이 연구해왔던 topic이다. 최근에는 기존의 gene based classifier 에서 탈피, pathway based classifier 로 진화하여 gene based 방법에 비해 좀 더 견고하고 정확도 높은 classifier 구축이 가능해졌다.
이 분야 선도 그룹은 UCSD의 Trey Ideker 그룹으로 KAIST의 이도헌 교수 연구실과 공동 연구를 통해 최근 2편의 pathway based classifier 논문을 출판하였다. ( Mole. Syst. Biol. , 3, 140 , Plos Comput. Biol. , 4, e1000217 )
기본적으로 이러한 Pathway 기반 disease classifier 연구의 정확도가 상대적으로 높은 이유는 single gene 레벨의 높은 발현 variation 이 pathway level 의 발현도로 summarize 되면서 상대적으로 발현의 variation이 낮아지기 때문인 것으로 판단된다.
예를 들어 Pathway A의 유전자 5개에서의 발현도가 각각
a1=10,a2=15,a3=30,a4=20,a5=50
이라 한다면 이 Pathway A의 평균 발현도는 25가 된다.
다른 실험에서 이 pathway A의 유전자 5개의 발현도가
a1=20,a2=30,a3=50,a4=30,a5=10
이라 한다면 이 실험 환경에서 pathway A의 평균 발현도는 28로 이전의 실험환경에서 얻어진 pathway 평균 발현도와 유사한 값을 나타내지만, 각각의 유전자 발현의 차이를 계산하면,
발현도 95( Euclidian distance )의 차이를 나타낸다.
오늘 Plos ONE 에 Pathway 기반 classifier 의 정확도를 향상시킨 논문이 출판되었다. ( Plos ONE, 4, e8161 ) 이 논문이 개선한 기존의 Pathway 기반 모델의 문제점은 Pathway 레벨의 발현도를 계산할 때, 하나의 Pathway에 속한 모든 유전자들의 발현도를 평균을 내어, Pathway에 속한 특정 유전자와 특정 phenotype 과의 관계를 정확히 반영하지 못한다는 점이다.
어떤 유전자는 특정 phenotype과 positive correlation 관계를, 어떤 유전자는 negative correlation 관계를 가질 수 있는데 Pathway 레벨로 모든 유전자들의 발현도가 평균되면
이러한 개개의 유전자와 phenotype의 관계가 사라져버리고, 전체 pathway 발현과
phenotype과의 관계만이 존재하게 된다. 물론 이렇게 되면서 개개의 유전자들의 발현 variation이 낮아져 상대적으로 안정적이고 재현도 높은 pathway 레벨의 feature가 생성되어 유전자 기반 모델보다 우수한 classification 정확도를 높이게 된 결정적인 원인이기도 하지만 말이다.
논문에서는 이런 문제점을 해결하고자 Pathway level의 발현도로 개개의 유전자 발현도를 summarize 할 때, 각 phenotype 그룹의 유전자 분포를 이용하여 특정 유전자의 서로 다른 두 phenotype 간의 PDF ( Probability Density Function ) 의 차이를 Log ratio 로 계산하여 각 유전자의 두 phenotype 간 발현도의 상대적 분포의 차를 구하여, 전체 pathway 의 phenotype discrimination score 를 구한다. Phenotype discrimination score 의 순위로 top feature 를 선정하고, 이를 바탕으로 phenotype classification 을 수행하게 되는데, 이 과정은 여타의 방법과 큰 차이가 없다.
여타의 Pathway classifier 와 비교하면 새로운 pathway scoring 방법이 우수한 성능을 보이는데, 정확도의 향상은 모든 test에서 5% 이하로 미미하긴 하나, 모든 test 에서 기존의 모든 방법론보다 우수한 결과를 나타내었다는 것은 유전자 context 기반 pathway scoring 이 기존의 pathway averaging 에 비해 pathway 기반 classifier 의 feature selection 방법으로 우수하다는 것을 보여준다 할 수 있다.
이 분야 선도 그룹은 UCSD의 Trey Ideker 그룹으로 KAIST의 이도헌 교수 연구실과 공동 연구를 통해 최근 2편의 pathway based classifier 논문을 출판하였다. ( Mole. Syst. Biol. , 3, 140 , Plos Comput. Biol. , 4, e1000217 )
기본적으로 이러한 Pathway 기반 disease classifier 연구의 정확도가 상대적으로 높은 이유는 single gene 레벨의 높은 발현 variation 이 pathway level 의 발현도로 summarize 되면서 상대적으로 발현의 variation이 낮아지기 때문인 것으로 판단된다.
예를 들어 Pathway A의 유전자 5개에서의 발현도가 각각
a1=10,a2=15,a3=30,a4=20,a5=50
이라 한다면 이 Pathway A의 평균 발현도는 25가 된다.
다른 실험에서 이 pathway A의 유전자 5개의 발현도가
a1=20,a2=30,a3=50,a4=30,a5=10
이라 한다면 이 실험 환경에서 pathway A의 평균 발현도는 28로 이전의 실험환경에서 얻어진 pathway 평균 발현도와 유사한 값을 나타내지만, 각각의 유전자 발현의 차이를 계산하면,
발현도 95( Euclidian distance )의 차이를 나타낸다.
오늘 Plos ONE 에 Pathway 기반 classifier 의 정확도를 향상시킨 논문이 출판되었다. ( Plos ONE, 4, e8161 ) 이 논문이 개선한 기존의 Pathway 기반 모델의 문제점은 Pathway 레벨의 발현도를 계산할 때, 하나의 Pathway에 속한 모든 유전자들의 발현도를 평균을 내어, Pathway에 속한 특정 유전자와 특정 phenotype 과의 관계를 정확히 반영하지 못한다는 점이다.
어떤 유전자는 특정 phenotype과 positive correlation 관계를, 어떤 유전자는 negative correlation 관계를 가질 수 있는데 Pathway 레벨로 모든 유전자들의 발현도가 평균되면
이러한 개개의 유전자와 phenotype의 관계가 사라져버리고, 전체 pathway 발현과
phenotype과의 관계만이 존재하게 된다. 물론 이렇게 되면서 개개의 유전자들의 발현 variation이 낮아져 상대적으로 안정적이고 재현도 높은 pathway 레벨의 feature가 생성되어 유전자 기반 모델보다 우수한 classification 정확도를 높이게 된 결정적인 원인이기도 하지만 말이다.
논문에서는 이런 문제점을 해결하고자 Pathway level의 발현도로 개개의 유전자 발현도를 summarize 할 때, 각 phenotype 그룹의 유전자 분포를 이용하여 특정 유전자의 서로 다른 두 phenotype 간의 PDF ( Probability Density Function ) 의 차이를 Log ratio 로 계산하여 각 유전자의 두 phenotype 간 발현도의 상대적 분포의 차를 구하여, 전체 pathway 의 phenotype discrimination score 를 구한다. Phenotype discrimination score 의 순위로 top feature 를 선정하고, 이를 바탕으로 phenotype classification 을 수행하게 되는데, 이 과정은 여타의 방법과 큰 차이가 없다.
여타의 Pathway classifier 와 비교하면 새로운 pathway scoring 방법이 우수한 성능을 보이는데, 정확도의 향상은 모든 test에서 5% 이하로 미미하긴 하나, 모든 test 에서 기존의 모든 방법론보다 우수한 결과를 나타내었다는 것은 유전자 context 기반 pathway scoring 이 기존의 pathway averaging 에 비해 pathway 기반 classifier 의 feature selection 방법으로 우수하다는 것을 보여준다 할 수 있다.
Sunday, December 6, 2009
Cross-ID mapping 문제 해결? BioGSP
Web address : http://biogps.gnf.org/
Bioinformatics 연구에서 다양한 소스의 데이터를 통합해야 하는 경우가
빈번하다. 유전자 이름만 해도 NCBI 내에서만 Gene ID, Entrez ID, Genbank ID
등등 다양하고, 기관 마다 차이가 존재하기 때문에, 이러한 다양한 기관의
ID 들 간의 cross-mapping 은 필수적이다.
그러나 매번 같은 소소의 정보를 이용하지 않는 경우가 많다보니, 매번 서로다른
기관들의 ID 매핑이 어려운 과정은 아니지만 반복되어 짜증을 유발하는 경우가
많다.

이번에 Genome Biology 에 소개된 ( http://genomebiology.com/2009/10/11/R130 )
BioGPS 는 이러한 cross-mapping 문제 해결을 위한 web-server 다.
BioGPS 에서 커버하는 ID 소스는
* Gene symbol ( from various institution )
* GO
* Interpro
* Affymetrix ID
등이고, primary ID mapping 파일을 다운로드 받을 수 있게 제공하고 있다.
Bioinformatics 연구에서 다양한 소스의 데이터를 통합해야 하는 경우가
빈번하다. 유전자 이름만 해도 NCBI 내에서만 Gene ID, Entrez ID, Genbank ID
등등 다양하고, 기관 마다 차이가 존재하기 때문에, 이러한 다양한 기관의
ID 들 간의 cross-mapping 은 필수적이다.
그러나 매번 같은 소소의 정보를 이용하지 않는 경우가 많다보니, 매번 서로다른
기관들의 ID 매핑이 어려운 과정은 아니지만 반복되어 짜증을 유발하는 경우가
많다.
이번에 Genome Biology 에 소개된 ( http://genomebiology.com/2009/10/11/R130 )
BioGPS 는 이러한 cross-mapping 문제 해결을 위한 web-server 다.
BioGPS 에서 커버하는 ID 소스는
* Gene symbol ( from various institution )
* GO
* Interpro
* Affymetrix ID
등이고, primary ID mapping 파일을 다운로드 받을 수 있게 제공하고 있다.
Tuesday, October 27, 2009
DEG selection 방법 어떤 걸 써야 할까? [ Comparison among nonparametric DEG selection methods ]
초창기 Microarray 연구에서 가장 큰 관심사 중 하나는 어떻게 하면 robust한 DEG ( Differentially Expressed Gene ) 을 찾아낼 수 있는가 였다.
가장 먼저 시도되었던 기본적인 방법들이 fold change 비교와 t-test 였는데, 이는 지금까지도 microarray data 분석의 가장 기본적인 방법으로 널리 이용되고 있다. 또한 MAQC( MicroArray Quality Control) 그룹의 보고에 따르면, 이 두가지 방법을 느슨한 stringency 를 주어 DEG를 뽑을 때, cross-platform, cross-laboratory microarray data에서 가장 재연성있고 안정정으로 DEG 셋을 얻을 수 있다고 하니, 가장 기본적이면서 또한 가장 중요한 DEG selection 방법이라고 할 수 있다.
이렇듯 가장 기본적인 DEG selection 방법인 t-test에도 약점이 있었으니, 그것은 t-test가 normal distribution을 가정한 parametric test라는 것이다. microarray data는 normal distribution을 따르지 않는 경우가 대부분이라고 할 수 있다. 자연스럽게 t-test의 이런 약점을 보완할 수 있는 방법들이 DEG selection 방법으로 적용되었는데, 대표적으로 non-parametric t-test와 rank sum test 를 들 수 있다.
오늘 이 posting 을 맘먹게 한 논문 ,Nonparametric identification methods for differentially expressed genes, bioinformatics, 18, 1454, 은 현재 Princeton 대학에서 교수로 재직중인 Olga G. Troyanskaya 가 Stanford 에서 박사과정 중 출판한 논문으로, 앞서 언급한 microarray data의 DEG selection methods 들 중, nonparametric method 3가지를 비교 분석한 논문이다.
논문에서 비교 분석 대상이 된 Non-parametric method 3가지는 non-parametric t-test, Wilcoxon rank sum test, Ideal discriminator method 되겠다. 이 3가지 방법을 simulated data 셋에 적용하여 TPR, FPR 을 측정하여 상대적인 비교우위에 대한 분석을 논하고 있는데, 결론부터 얘기하자면, Rank sum test 를 쓰는 것이 다른 방법들에 비해 stringent 한 결과를 주고, multiple permutation 을 통한 significance 를 측정할 필요가 없기 때문에 일반적인 상황에서는 rank sum test를 쓰는 것이 이 세가지 방법 중 가장 computationally efficient 하고, 상대적으로 보수적이지만 정확하게 DEG set 을 뽑아낼 수 있다는 것. 덧붙여 p-value 0.1 정도에서 sensitivity와 specificity 의 trade-off 에서 가장 만족스러운 결과를 얻을 수 있었다는 결과가 리포팅 되어 있다.
차후에 SAM,GSEA 등을 비롯한 다양한 DEG selection 방법이 소개되었으나, 기본적인 single gene based DEG selection analysis 는 기본적으로 필요한 과정이니, microarray 분석을 할 때 기 이를 염두에 두고 nonparametric test 를 적용해보는 것이 좋을 듯 하다.
이 논문이 출판되고 4년 후, MAQC 그룹이 출판한 Nature 논문 에 따르면 느슨한 t-test p-value 0.1 와 fold change 1.2~1.5 정도를 함께 사용하여 DEG set 을 선정하였을 때, 가장 만족스러운 결과를 얻을 수 있었다고 보고하고 있으니, 이 결과도 참고해둘 만 하다.
* Non-parametric t-test : normal distribution 을 가정한 상태로 p-value를 구하지 않고, permutation test에 의해 t-test statistics 의 distribution 을 만들고 이와 원 t-test statistics 값을 비교하여 p-value significance 를 구한다.
가장 먼저 시도되었던 기본적인 방법들이 fold change 비교와 t-test 였는데, 이는 지금까지도 microarray data 분석의 가장 기본적인 방법으로 널리 이용되고 있다. 또한 MAQC( MicroArray Quality Control) 그룹의 보고에 따르면, 이 두가지 방법을 느슨한 stringency 를 주어 DEG를 뽑을 때, cross-platform, cross-laboratory microarray data에서 가장 재연성있고 안정정으로 DEG 셋을 얻을 수 있다고 하니, 가장 기본적이면서 또한 가장 중요한 DEG selection 방법이라고 할 수 있다.
이렇듯 가장 기본적인 DEG selection 방법인 t-test에도 약점이 있었으니, 그것은 t-test가 normal distribution을 가정한 parametric test라는 것이다. microarray data는 normal distribution을 따르지 않는 경우가 대부분이라고 할 수 있다. 자연스럽게 t-test의 이런 약점을 보완할 수 있는 방법들이 DEG selection 방법으로 적용되었는데, 대표적으로 non-parametric t-test와 rank sum test 를 들 수 있다.
오늘 이 posting 을 맘먹게 한 논문 ,Nonparametric identification methods for differentially expressed genes, bioinformatics, 18, 1454, 은 현재 Princeton 대학에서 교수로 재직중인 Olga G. Troyanskaya 가 Stanford 에서 박사과정 중 출판한 논문으로, 앞서 언급한 microarray data의 DEG selection methods 들 중, nonparametric method 3가지를 비교 분석한 논문이다.
논문에서 비교 분석 대상이 된 Non-parametric method 3가지는 non-parametric t-test, Wilcoxon rank sum test, Ideal discriminator method 되겠다. 이 3가지 방법을 simulated data 셋에 적용하여 TPR, FPR 을 측정하여 상대적인 비교우위에 대한 분석을 논하고 있는데, 결론부터 얘기하자면, Rank sum test 를 쓰는 것이 다른 방법들에 비해 stringent 한 결과를 주고, multiple permutation 을 통한 significance 를 측정할 필요가 없기 때문에 일반적인 상황에서는 rank sum test를 쓰는 것이 이 세가지 방법 중 가장 computationally efficient 하고, 상대적으로 보수적이지만 정확하게 DEG set 을 뽑아낼 수 있다는 것. 덧붙여 p-value 0.1 정도에서 sensitivity와 specificity 의 trade-off 에서 가장 만족스러운 결과를 얻을 수 있었다는 결과가 리포팅 되어 있다.
차후에 SAM,GSEA 등을 비롯한 다양한 DEG selection 방법이 소개되었으나, 기본적인 single gene based DEG selection analysis 는 기본적으로 필요한 과정이니, microarray 분석을 할 때 기 이를 염두에 두고 nonparametric test 를 적용해보는 것이 좋을 듯 하다.
이 논문이 출판되고 4년 후, MAQC 그룹이 출판한 Nature 논문 에 따르면 느슨한 t-test p-value 0.1 와 fold change 1.2~1.5 정도를 함께 사용하여 DEG set 을 선정하였을 때, 가장 만족스러운 결과를 얻을 수 있었다고 보고하고 있으니, 이 결과도 참고해둘 만 하다.
* Non-parametric t-test : normal distribution 을 가정한 상태로 p-value를 구하지 않고, permutation test에 의해 t-test statistics 의 distribution 을 만들고 이와 원 t-test statistics 값을 비교하여 p-value significance 를 구한다.
Wednesday, March 18, 2009
BioTool::NCBIfetch
Nowadays, I've tried to build my own perl pacakges selections on routine bioinformatics tasks.
Althrough BioPerl is already out there, it usually doesn't have modules for my own purpose.
For example, a package for learning PAML packages from A to Z, not requiring specific formatting to run the BioPerl. Well, so I started to build my own perl packages.
I named the base package name 'BioTool' and added packages whenever I confronted any situation
that I found some tasks might be used repeatly and routinely.
Recenlty, I made 'BioTool::NCBIfetch' package, which can extract sequences and all the related references db information including Gene symbol, Gene description, Chromosome location, Ensembl, Unigene, Uniprot, KEGG and GO for a given gene query.
Since the package fetches the information from result of NCBI eutils query, which results with up-to-date information, the analysis result for a given gene is up-to-dated. So users don't need to worried about whether the reference data is out-dated when they use a program which works the same taks based on localized data. In short, users could free themselves from updating all related indenpent databases day to day.
I'll show some example usage of this package
This package might be very helpful for ones who want to cross mapping between major biology databases based on NCBI gene identifier. I actually made it for cross-mapping genes in GEO platform since GEO platform annotation is frequently incomplete and inaccurate.
If anyone interests in this pacakge and want to use, request!
Althrough BioPerl is already out there, it usually doesn't have modules for my own purpose.
For example, a package for learning PAML packages from A to Z, not requiring specific formatting to run the BioPerl. Well, so I started to build my own perl packages.
I named the base package name 'BioTool' and added packages whenever I confronted any situation
that I found some tasks might be used repeatly and routinely.
Recenlty, I made 'BioTool::NCBIfetch' package, which can extract sequences and all the related references db information including Gene symbol, Gene description, Chromosome location, Ensembl, Unigene, Uniprot, KEGG and GO for a given gene query.
Since the package fetches the information from result of NCBI eutils query, which results with up-to-date information, the analysis result for a given gene is up-to-dated. So users don't need to worried about whether the reference data is out-dated when they use a program which works the same taks based on localized data. In short, users could free themselves from updating all related indenpent databases day to day.
I'll show some example usage of this package
use BioTool::NCBIfetch;
my $ncbi=BioTool::NCBIfetch->new;
$fetch->set_query(gene,780,xml); # Input : search DB, ID(geneID, gID), result type, result format type)
$fetch->get_result;
my $symbol=$fetch->get_symbol;
my @TreEMBL=$fetch->get_uniprotTreEMBL;
print "$symbol\t@TreEMBL\n";
This package might be very helpful for ones who want to cross mapping between major biology databases based on NCBI gene identifier. I actually made it for cross-mapping genes in GEO platform since GEO platform annotation is frequently incomplete and inaccurate.
If anyone interests in this pacakge and want to use, request!
Sunday, January 4, 2009
CRONOS
Web address : http://mips.gsf.de/genre/proj/cronos/index.html
Protein-protein interaction DB로 널리 알려진 MIPS 에서
gene과 protein 의 major 관리 기관의 ID 들 간의 cross-mapping
정보를 담은 DB를 공개했다.
NCBI의 refseq ID와 UniprotKB, Ensembl 이 3개 major ID 소스로
이용이 되었고, 이 외에도 PIR 등의 DB, 나아가 affymetrix 의 칩 ID와도
cross-mapping web-service를 제공한다.
Protein-protein interaction DB로 널리 알려진 MIPS 에서
gene과 protein 의 major 관리 기관의 ID 들 간의 cross-mapping
정보를 담은 DB를 공개했다.
NCBI의 refseq ID와 UniprotKB, Ensembl 이 3개 major ID 소스로
이용이 되었고, 이 외에도 PIR 등의 DB, 나아가 affymetrix 의 칩 ID와도
cross-mapping web-service를 제공한다.
Thursday, December 18, 2008
Mapping Ensembl to external
Bio data들 간의 Cross-mapping 은 어떤 데이터를 다루든 맨처음 해결해야 하는 문제로, 때때로 골머리를 썩히곤 한다.
Ensembl의 경우 Ensembl 고유의 gene, protein 에 대한 ID를 extermal bio database 들과 mapping 시켜주는 서비스를
Biomart라는 툴을 통해 제공하고 있다.
Biomart at Ensembl : http://www.ensembl.org/biomart
사용법은 위의 url 에 연결된 biomart 홈페이지에 가서 원하는 ensembl genome data와 필요한 external database를
선택하고, 결과 파일을 다운 받으면 된다.
Ensembl의 경우 Ensembl 고유의 gene, protein 에 대한 ID를 extermal bio database 들과 mapping 시켜주는 서비스를
Biomart라는 툴을 통해 제공하고 있다.
Biomart at Ensembl : http://www.ensembl.org/biomart
사용법은 위의 url 에 연결된 biomart 홈페이지에 가서 원하는 ensembl genome data와 필요한 external database를
선택하고, 결과 파일을 다운 받으면 된다.
Tuesday, December 16, 2008
Pairwise gene-gene co-expression 계산 시간 단축
몇일전 Affymetrix chip 의 모든 probeset vs. probeset correlation coefficient 계산을 CPAN 모듈을 이용해서 수행했다. 계산량이 45001*45000/2 , 10억 번이 넘는 어마어마한 양이라 지레 짐작으로 꾀나 걸리겠구나 하고 그냥 두었는데 하루가 지나서 보니 2.2GB 용량에 기껏 몇천만개 정도만 계산이 된 상태. 썅.
부랴부랴 계산 시간을 점검해 봤더니 하나당 0.003초 정도.
전체 계산 횟수 45001*45000/2 곱하기 0.003 초. 이걸 다시 3600초로 나눠 몇시간이나 드는가를 계산해 보면 843.7687 시간, 35일은 더 돌려야 하는 엄청난 계산 시간!
CPAN 모듈을 쓴 경우 probeset pair에 대한 correlation 계산이 각각에 대한 standard deviation을 매번 계산하기 때문에, 중복된 계산이 어마어마하다. 그래서 먼저 각 probeset 의 expression data를 z-score로 transform 했다. z-score transform 된 경우 두 probeset의 z-score transformed expression vector를 곱해서 더해주기면 하면 correlation coefficient가 되기 때문에 계산량이 대폭 줄어든다. (포스팅 참조)
이렇게 한 결과 하나당 계산 시간이 0.0001초로, 약 30배로 계산 속도가 빨라졌다.
이렇게 해도 전체 셋에 대한 계산은 44시간이 걸리긴 한다만...
부랴부랴 계산 시간을 점검해 봤더니 하나당 0.003초 정도.
전체 계산 횟수 45001*45000/2 곱하기 0.003 초. 이걸 다시 3600초로 나눠 몇시간이나 드는가를 계산해 보면 843.7687 시간, 35일은 더 돌려야 하는 엄청난 계산 시간!
CPAN 모듈을 쓴 경우 probeset pair에 대한 correlation 계산이 각각에 대한 standard deviation을 매번 계산하기 때문에, 중복된 계산이 어마어마하다. 그래서 먼저 각 probeset 의 expression data를 z-score로 transform 했다. z-score transform 된 경우 두 probeset의 z-score transformed expression vector를 곱해서 더해주기면 하면 correlation coefficient가 되기 때문에 계산량이 대폭 줄어든다. (포스팅 참조)
## Correlation function 이용한 경우
correlation(\@pair1,\@pair2 );
sub correlation{
my $i=shift;
my $j=shift;
my $sd1=standardize($i);
my $sd2=standardize($j);
my $cor;
$cor+=$sd1->[$_]*$sd2->[$_] for 0..$#$sd1;
$cor/=$#$sd1;
return $cor;
}
## 먼저 z-score transform 한 경우
$cor+=$pair1[$_]*$pair2[$_] for 0..$#pair1;
$cor/=$#pair1;
이렇게 한 결과 하나당 계산 시간이 0.0001초로, 약 30배로 계산 속도가 빨라졌다.
이렇게 해도 전체 셋에 대한 계산은 44시간이 걸리긴 한다만...
Wednesday, January 9, 2008
Integrating gene expression data with other biological data
Microarray data 를 이용한 연구가 많이 이뤄져왔고, 이루어지고 있지만, 기술적인 한계 or 생물학적 시스템의 불안정성으로 인해 각 데이터 간의 재연성이 높지 않고, 따라서 신뢰도가 떨어지는 실정이다. 생물학 시스템이 정확히 하나의 expression pattern 그대로 있는 시간은 얼마나 될까? Microarray 실험을 하기 위한 sample을 얻는 시간을 컨트롤하여 정확하게 같은 expression pattern을 재연할 수 있을까?
똑같은 Pancreatic cancer cell에 대한 expression pattern도 cell을 둘러싼 환경에 따라, 시간에 따라 달라진다. 서로 다른 환자에게서 추출된 cancer cell 간의 pattern 이라면 영향을 끼치는 factor들은 더욱 많아지게 된다. Gene expression pattern의 재연성이 떨어지는 것은 이러한 정황을 고려해 볼 때 당연한 결과인지 모른다.
Expression pattern을 통해 목적하는 일 중 대부분이 Biomarker selection 이다. Lung cancer를 일으키는 gene은? Diabetis를 일으키는 gene은? 이런 식으로 찾아낸 gene을 가지고 drug target을 삼고, drug discovery에 돌입하는 traditional research paradigm 이 궁극적인 목적이 된다.
그러나 아이러니하게 gene expression data는 오히려 이런 old paradigm 보다는, 전체적인 expression 양상, 단일한 gene이 아닌 전체적인 gene들의 발현 양상이 서로 어떻게 연관되어 있는지를 연구할 수 있는 new paradigm에 적합하다.
Protein interaction, gene interaction, coexpression analysis 등과의 접목을 통해 expression data를 1차원에서 2차원, 3차원으로 높여 분석할 때, 이런 Global, Local gene expression pattern 뒤에 감춰진 의미를 더욱 잘 이해할 수 있을 것이다.
똑같은 Pancreatic cancer cell에 대한 expression pattern도 cell을 둘러싼 환경에 따라, 시간에 따라 달라진다. 서로 다른 환자에게서 추출된 cancer cell 간의 pattern 이라면 영향을 끼치는 factor들은 더욱 많아지게 된다. Gene expression pattern의 재연성이 떨어지는 것은 이러한 정황을 고려해 볼 때 당연한 결과인지 모른다.
Expression pattern을 통해 목적하는 일 중 대부분이 Biomarker selection 이다. Lung cancer를 일으키는 gene은? Diabetis를 일으키는 gene은? 이런 식으로 찾아낸 gene을 가지고 drug target을 삼고, drug discovery에 돌입하는 traditional research paradigm 이 궁극적인 목적이 된다.
그러나 아이러니하게 gene expression data는 오히려 이런 old paradigm 보다는, 전체적인 expression 양상, 단일한 gene이 아닌 전체적인 gene들의 발현 양상이 서로 어떻게 연관되어 있는지를 연구할 수 있는 new paradigm에 적합하다.
Protein interaction, gene interaction, coexpression analysis 등과의 접목을 통해 expression data를 1차원에서 2차원, 3차원으로 높여 분석할 때, 이런 Global, Local gene expression pattern 뒤에 감춰진 의미를 더욱 잘 이해할 수 있을 것이다.
Wednesday, November 21, 2007
Fun&Co
Article source : Bioinformatics, 23, 2725, 2007
두 그룹의 microarray data set의 functional difference를
비교하고자 할 때 ( e.g. 서로 다른 tissue, disease vs. normal 등)
사용할 수 있는 방법으로 fun&co 이라는 web server가 공개되었다.
Procedure는 아래와 같다.
1. 각 그룹의 intensity를 rank로 변환
2. 가능한 모든pair probe set에 대해 spearman correlation coefficient 계산
3. pair probe set이 동일한 gene을 detection하는 경우가 아닐 때, 연관된 GOmain term을 counting
4. 각 그룹의 counting 된 GO term set을 비교하여, 그룹과 Over-correlated or under-correlated 된 GO 를 선정
다른 방법들과 구분되는 특이한 점이라면,
하나의 GO term을 지칭하는 Pair gene이
correlation되는 경우 연관된 GO term에만
의미를 두었다는 것이다.
이는 multi data set comparision의 경우 몇가지 advantage를 가지는데
1. 각 data set에서 DEG test를 할 필요가 없다는 점
2. 따라서 normal condition에 대한 고려가 필요없는 경우 ( 서로 다른 tissue 비교와 같은 경우), 각 data set에서 관련된 array만 고려하면 된다는 점
등이 된다.
두 그룹의 microarray data set의 functional difference를
비교하고자 할 때 ( e.g. 서로 다른 tissue, disease vs. normal 등)
사용할 수 있는 방법으로 fun&co 이라는 web server가 공개되었다.
Procedure는 아래와 같다.
1. 각 그룹의 intensity를 rank로 변환
2. 가능한 모든pair probe set에 대해 spearman correlation coefficient 계산
3. pair probe set이 동일한 gene을 detection하는 경우가 아닐 때, 연관된 GOmain term을 counting
4. 각 그룹의 counting 된 GO term set을 비교하여, 그룹과 Over-correlated or under-correlated 된 GO 를 선정
다른 방법들과 구분되는 특이한 점이라면,
하나의 GO term을 지칭하는 Pair gene이
correlation되는 경우 연관된 GO term에만
의미를 두었다는 것이다.
이는 multi data set comparision의 경우 몇가지 advantage를 가지는데
1. 각 data set에서 DEG test를 할 필요가 없다는 점
2. 따라서 normal condition에 대한 고려가 필요없는 경우 ( 서로 다른 tissue 비교와 같은 경우), 각 data set에서 관련된 array만 고려하면 된다는 점
등이 된다.
Tuesday, November 20, 2007
Human PAML Brower
Human gene들에 대한 evolutionary rate을 PAML 패키지를
이용해 계산한 결과를 담은 DB가 공개 되었다.
Human PAML Browser : http://mendel.gene.cwru.edu/adamslab/pbrowser.py
Human gene은 약 22,000~28,000 로 추정되는데,
여기서는 UCSC multispecies genome alignment에
의해 얻어진 ortholog 13,721개에 대한 결과를 담고있다.
PAML을 돌리기 위해 DNA sequence pair, Protein sequence pair
들을 각각 준비해야 하고, computation time도 꾀나 걸린다는
점을 생각해 볼 때, 필요할 때 마다 관심있는 gene의 PAML
결과를 검색할 때 유용할 것 같다.
NAR paper : http://nar.oxfordjournals.org/cgi/reprint/gkm764v1?ck=nck
이용해 계산한 결과를 담은 DB가 공개 되었다.
Human PAML Browser : http://mendel.gene.cwru.edu/adamslab/pbrowser.py
Human gene은 약 22,000~28,000 로 추정되는데,
여기서는 UCSC multispecies genome alignment에
의해 얻어진 ortholog 13,721개에 대한 결과를 담고있다.
PAML을 돌리기 위해 DNA sequence pair, Protein sequence pair
들을 각각 준비해야 하고, computation time도 꾀나 걸린다는
점을 생각해 볼 때, 필요할 때 마다 관심있는 gene의 PAML
결과를 검색할 때 유용할 것 같다.
NAR paper : http://nar.oxfordjournals.org/cgi/reprint/gkm764v1?ck=nck
Subscribe to:
Posts (Atom)